From single scattering to multiple scattering effects¶
In this tutorial we will observe one of the main difference between a single and a multiple scatteirng process: the defocusing effect.
We will reproduce a calculation performed by M.-L Xu, J.J. Barton and M.A. Van Hove in their paper of 1989 (see below ) In the spirit of figure 3 of their paper, We create 3 atomic chains of Ni atoms (2, 3 and 5 atoms) tilted by 45° and we compare the intensity of the forward scattering peak as a function of the scattering order: for single scattering and for a multiple scattering at the 5th order.
Below is a commented version of this example. You can download it here
1# coding: utf-8
2
3# import all we need and start by msspec
4from msspec.calculator import MSSPEC
5
6# we will build a simple atomic chain
7from ase import Atom, Atoms
8
9# we need some numpy functions
10import numpy as np
11
12
13symbol = 'Ni' # The kind of atom for the chain
14orders = (1, 5) # We will run the calculation for single scattering
15 # and multiple scattering (5th diffusion order)
16chain_lengths = (2,3,5) # We will run the calculation for differnt lengths
17 # of the atomic chain
18a = 3.499 * np.sqrt(2)/2 # The distance bewteen 2 atoms
19
20# Define an empty variable to store all the results
21all_data = None
22
23# 2 for nested loops over the chain length and the order of diffusion
24for chain_length in chain_lengths:
25 for order in orders:
26 # We build the atomic chain by
27 # 1- stacking each atom one by one along the z axis
28 chain = Atoms([Atom(symbol, position = (0., 0., i*a)) for i in
29 range(chain_length)])
30 # 2- rotating the chain by 45 degrees with respect to the y axis
31 #chain.rotate('y', np.radians(45.))
32 chain.rotate(45., 'y')
33 # 3- setting a custom Muffin-tin radius of 1.5 angstroms for all
34 # atoms (needed if you want to enlarge the distance between
35 # the atoms while keeping the radius constant)
36 #[atom.set('mt_radius', 1.5) for atom in chain]
37 # 4- defining the absorber to be the first atom in the chain at
38 # x = y = z = 0
39 chain.absorber = 0
40
41 # We define a new PED calculator
42 calc = MSSPEC(spectroscopy = 'PED')
43 calc.set_atoms(chain)
44 # Here is how to tweak the scattering order
45 calc.calculation_parameters.scattering_order = order
46 # This line below is where we actually run the calculation
47 all_data = calc.get_theta_scan(level='3s', #kinetic_energy=1000.,
48 theta=np.arange(0., 80.), data=all_data)
49
50 # OPTIONAL, to improve the display of the data we will change the dataset
51 # default title as well as the plot title
52 t = "order {:d}, n = {:d}".format(order, chain_length) # A useful title
53 dset = all_data[-1] # get the last dataset
54 dset.title = t # change its title
55 # get its last view (there is only one defined for each dataset)
56 v = dset.views()[-1]
57 v.set_plot_options(title=t) # change the title of the figure
58
59
60
61# OPTIONAL, set the same scale for all plots
62# 1. iterate over all computed cross_sections to find the absolute minimum and
63# maximum of the data
64min_cs = max_cs = 0
65for dset in all_data:
66 min_cs = min(min_cs, np.min(dset.cross_section))
67 max_cs = max(max_cs, np.max(dset.cross_section))
68
69# 2. for each view in each dataset, change the scale accordingly
70for dset in all_data:
71 v = dset.views()[-1]
72 v.set_plot_options(ylim=[min_cs, max_cs])
73
74# Pop up the graphical window
75all_data.view()
76# You can end your script with the line below to remove the temporary
77# folder needed for the calculation
78calc.shutdown()
The resulting ouput is reported below. We added a sketch of the atomic chains on the left hand side of the figure. You can see that for the single scattering computation (left column), the forward peak is growing with increasing the number of atoms in the chain, while it is clearly damped when using the multiple scattering approach. Electrons are focused by the second atom in the chain and over focused again by the third atom leading to a diverging trajectory for this electron which in turn lowers the signal.
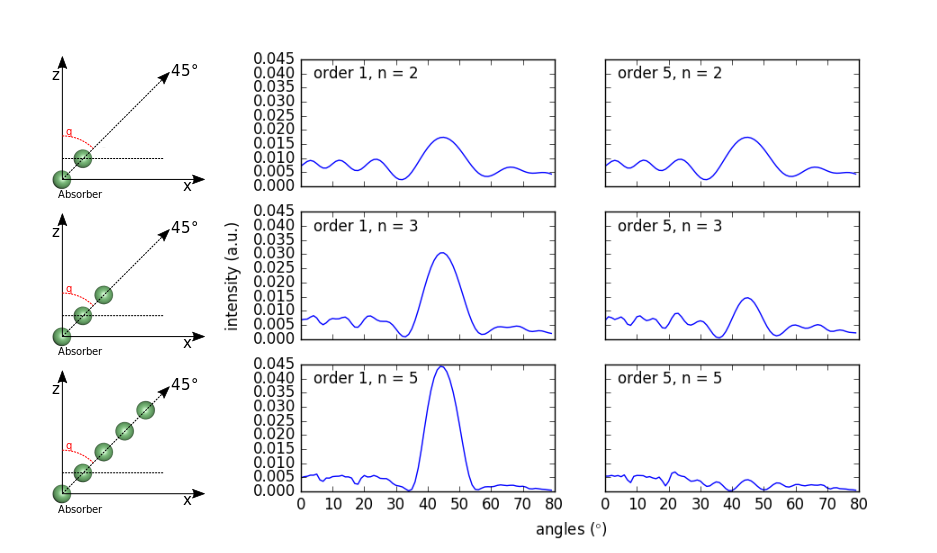
Figure 1. Polar scan of a Ni chain of 2-5 atoms for single and mutliple (5th order) scattering.¶
See also
- Electron scattering by atomic chains: Multiple-scattering effects
M.-L Xu, J.J. Barton & M.A. Van Hove, Phys. Rev. B 39 (12) p8275 (1989) [doi]